Designing Low Thermal Expansion Lattices at the Microscale
Research by John Chu
Objective
 Low thermal expansion materials are desired in the application of a mirror in a space telescope. Materials with a low coefficient of thermal expansion (CTE) are desired in order to minimize thermal stresses and strains caused by large variations in temperature and preserve the geometry of the optical surface. The concepts of low thermal expansion bi-material lattices are therefore used to create an optical surface which has a CTE of zero. In this application, the size of the unit cells must be on the order of 1 micron in length in order to avoid optical diffraction. At these small length scales, special considerations must be made. This research is conducted in collaboration with the Keck Institute for Space Studies (KISS) and Professor Chiara Daraio's group at the California Institute of Technology.
Low thermal expansion materials are desired in the application of a mirror in a space telescope. Materials with a low coefficient of thermal expansion (CTE) are desired in order to minimize thermal stresses and strains caused by large variations in temperature and preserve the geometry of the optical surface. The concepts of low thermal expansion bi-material lattices are therefore used to create an optical surface which has a CTE of zero. In this application, the size of the unit cells must be on the order of 1 micron in length in order to avoid optical diffraction. At these small length scales, special considerations must be made. This research is conducted in collaboration with the Keck Institute for Space Studies (KISS) and Professor Chiara Daraio's group at the California Institute of Technology.
|
Analytical and Numerical Study of Bi-Material Lattice
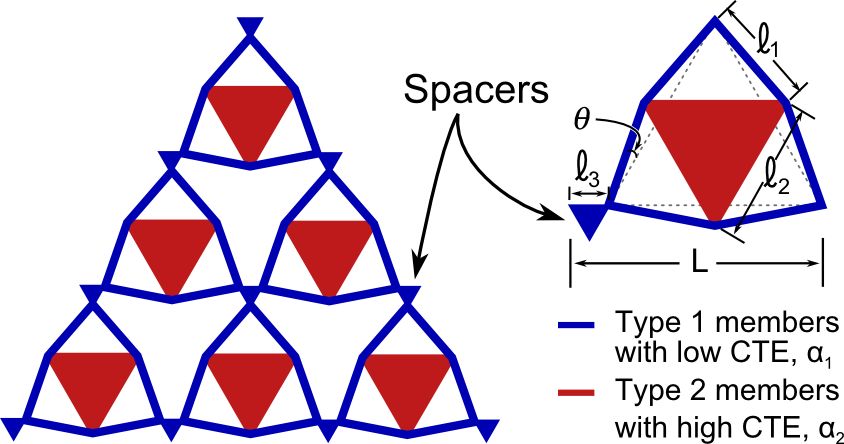 Features known as spacers have been incorporated into the unit cell geometry to avoid the formation of non-ideal lattice nodes. Non-ideal nodes are created by the intersection of constituent 1 members near the interface of adjacent unit cells and have the effect of increasing the overall CTE of the structure. The addition of spacers alleviates the problem of non-ideal lattice nodes and also facilitates larger skewness angles. To aid the design and development of the bi-material lattices, analytical models are developed from first principals to predict the thermal expansion of the new lattice configuration. The theoretical thermal expansion of the new design is derived under the assumption that the joints are bonded by constructing the global stiffness matrix of the unit cell. An analytical solution for the net CTE of the unit cell is therefore obtained by solving the nodal displacements of the structure given a change in temperature.
Features known as spacers have been incorporated into the unit cell geometry to avoid the formation of non-ideal lattice nodes. Non-ideal nodes are created by the intersection of constituent 1 members near the interface of adjacent unit cells and have the effect of increasing the overall CTE of the structure. The addition of spacers alleviates the problem of non-ideal lattice nodes and also facilitates larger skewness angles. To aid the design and development of the bi-material lattices, analytical models are developed from first principals to predict the thermal expansion of the new lattice configuration. The theoretical thermal expansion of the new design is derived under the assumption that the joints are bonded by constructing the global stiffness matrix of the unit cell. An analytical solution for the net CTE of the unit cell is therefore obtained by solving the nodal displacements of the structure given a change in temperature.
|
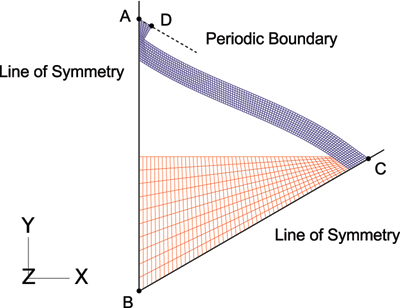 To validate the analytical expression derived in this work, finite element (FE) models are constructed in ABAQUS to simulate the thermal expansion of a unit cell. Aluminum (red) and titanium (blue) constituents are used in this analysis and are assumed to have isotropic and elastic material properties. Only one-sixth of the structure is modeled by taking advantage of symmetry and employing the appropriate boundary conditions. Nodes that lie on the lines of symmetry (AB and BC) are constrained to move along these lines, and the slope of the periodic boundary (AD) is kept constant. A bonded joint configuration is modeled by tying the degrees of freedom of the nodes at the material interface. As a result of thermal expansion, the titanium members will undergo bending due to the rotational resistance at the joints. The displacement of point A due to a change in temperature is therefore used to calculated the net CTE of the unit cell.
To validate the analytical expression derived in this work, finite element (FE) models are constructed in ABAQUS to simulate the thermal expansion of a unit cell. Aluminum (red) and titanium (blue) constituents are used in this analysis and are assumed to have isotropic and elastic material properties. Only one-sixth of the structure is modeled by taking advantage of symmetry and employing the appropriate boundary conditions. Nodes that lie on the lines of symmetry (AB and BC) are constrained to move along these lines, and the slope of the periodic boundary (AD) is kept constant. A bonded joint configuration is modeled by tying the degrees of freedom of the nodes at the material interface. As a result of thermal expansion, the titanium members will undergo bending due to the rotational resistance at the joints. The displacement of point A due to a change in temperature is therefore used to calculated the net CTE of the unit cell.
|
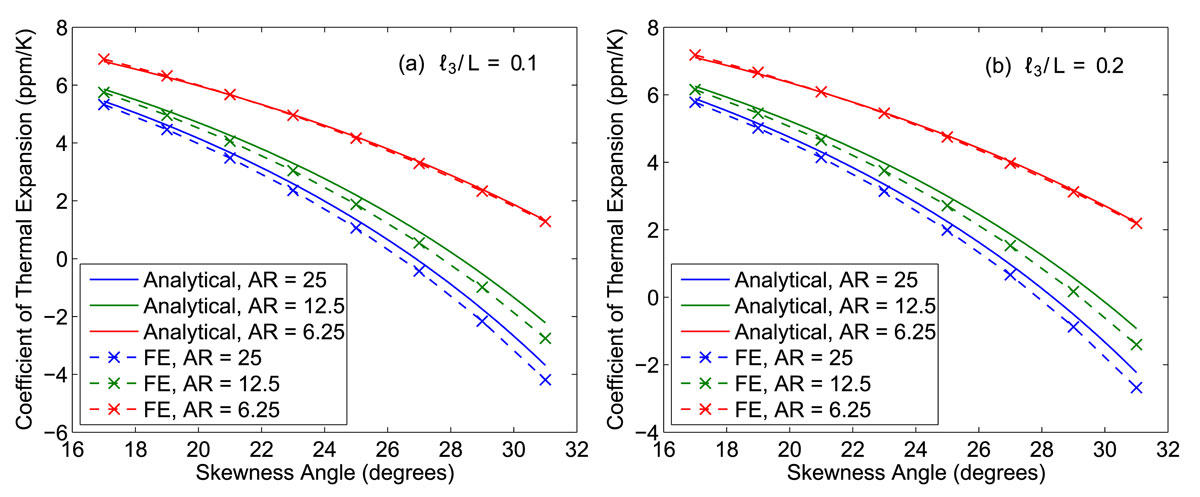 The thermal expansion of different unit cell configurations (thickness of type 1 constituents and size of spacer varied) is studied using FE models created in ABAQUS. Results from the numerical study are compared to the predictions given by the analytical expression derived for a bonded joint lattice. When plotted against each other, excellent agreement is found between theory and simulation for all unit cell geometries. Thus the analytical equation that was derived to predict the CTE of a given lattice configuration has been validated through numerical analysis.
The thermal expansion of different unit cell configurations (thickness of type 1 constituents and size of spacer varied) is studied using FE models created in ABAQUS. Results from the numerical study are compared to the predictions given by the analytical expression derived for a bonded joint lattice. When plotted against each other, excellent agreement is found between theory and simulation for all unit cell geometries. Thus the analytical equation that was derived to predict the CTE of a given lattice configuration has been validated through numerical analysis.
|
Molecular Dynamics Study of Amorphous Aluminum and Titanium
To manufacture the bi-material lattices at the microscale, thin films of aluminum and titanium created via electron-beam deposition are used. As a result of the deposition process, the atomic structure of the thin films has been determined to be amorphous through x-ray diffraction experiments. The thermal expansion and recrystallization of amorphous aluminum and titanium are not well documented in the literature, thus molecular dynamics (MD) simulations are conducted to study their material properties. All MD simulations performed in this research are done using LAMMPS (Large-scale Atomic/Molecular Massively Parallel Simulator), an open source parallel MD code developed and maintained by Sandia National Laboratories.
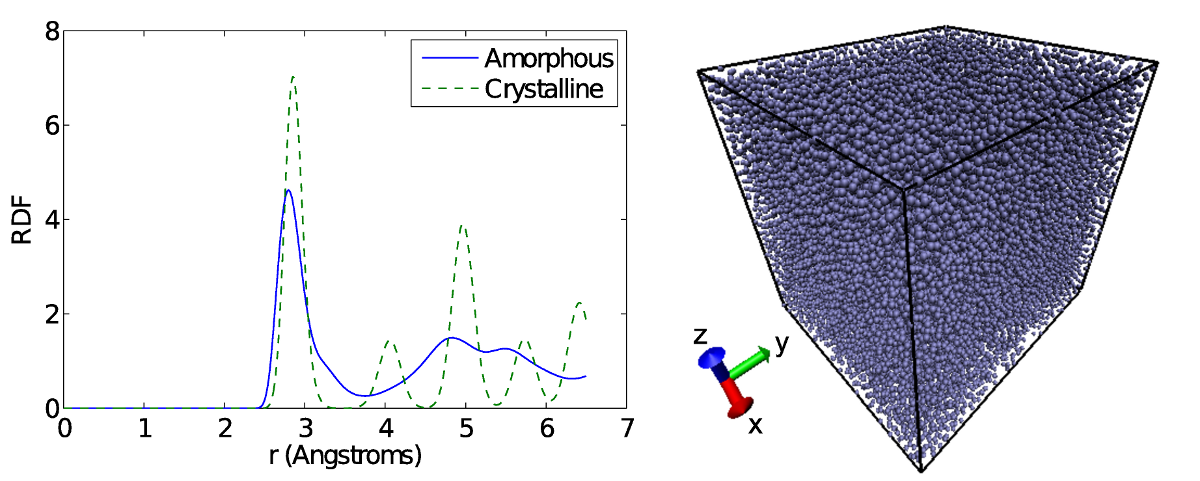 Amorphous states of aluminum and titanium are first created by rapidly quenching each material from their liquid state. Due to the rapid cooling process, the nucleation and growth of crystalline atomic structures is prevented and thus an energetically unfavourable amorphous state is obtained. To confirm the generation of amorphous materials, the radial distribution function (RDF) of the system is analyzed and the atomic coordinates are visualized using Visual Molecular Dynamics (VMD). The RDF measures from any given atom, the number of atoms found at a distance r normalized by the number that would be found in an equally distributed system. For a crystalline system, the RDF has multiple well defined peaks as the atoms sit and oscillate about their lattice positions. In an amorphous material, the first peak is shifted slightly to the left and the second peak is much broader than the first, with two apexes observed. The split second peak of the RDF is a common characteristic of amorphous materials. By visualizing the atoms of the system it is found that the atoms are in a disordered state, with no crystal lattices visible.
Amorphous states of aluminum and titanium are first created by rapidly quenching each material from their liquid state. Due to the rapid cooling process, the nucleation and growth of crystalline atomic structures is prevented and thus an energetically unfavourable amorphous state is obtained. To confirm the generation of amorphous materials, the radial distribution function (RDF) of the system is analyzed and the atomic coordinates are visualized using Visual Molecular Dynamics (VMD). The RDF measures from any given atom, the number of atoms found at a distance r normalized by the number that would be found in an equally distributed system. For a crystalline system, the RDF has multiple well defined peaks as the atoms sit and oscillate about their lattice positions. In an amorphous material, the first peak is shifted slightly to the left and the second peak is much broader than the first, with two apexes observed. The split second peak of the RDF is a common characteristic of amorphous materials. By visualizing the atoms of the system it is found that the atoms are in a disordered state, with no crystal lattices visible.
|
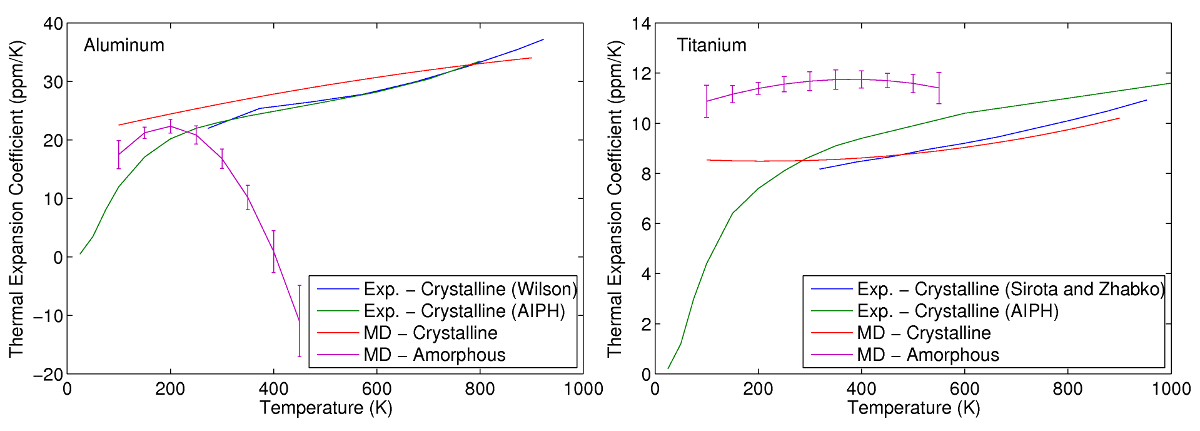 Via MD simulations, the thermal expansion of aluminum and titanium is determined by heating the simulation cell and measuring the changes in length. The CTEs of aluminum and titanium in both crystalline and amorphous states are studied to investigate the differences. Experimental data for crystalline aluminum and titanium are also plotted in comparison. Good agreement is found between the experimental and predicted CTEs of crystalline aluminum and titanium, illustrating the accuracy of the interatomic potentials chosen. The thermal expansion of both amorphous aluminum and titanium are found to differ from that of their crystalline counterparts. In the case of aluminum, the thermal expansion of the amorphous phase is comparable at temperatures below 250K, but drops off rapidly at higher temperatures. This is due to the onset of recrystallization in which the amorphous phase transforms into a denser crystalline state. For titanium, the CTE of its amorphous state is found to be consistently higher than its crystalline phase throughout the entire simulated temperature range.
Via MD simulations, the thermal expansion of aluminum and titanium is determined by heating the simulation cell and measuring the changes in length. The CTEs of aluminum and titanium in both crystalline and amorphous states are studied to investigate the differences. Experimental data for crystalline aluminum and titanium are also plotted in comparison. Good agreement is found between the experimental and predicted CTEs of crystalline aluminum and titanium, illustrating the accuracy of the interatomic potentials chosen. The thermal expansion of both amorphous aluminum and titanium are found to differ from that of their crystalline counterparts. In the case of aluminum, the thermal expansion of the amorphous phase is comparable at temperatures below 250K, but drops off rapidly at higher temperatures. This is due to the onset of recrystallization in which the amorphous phase transforms into a denser crystalline state. For titanium, the CTE of its amorphous state is found to be consistently higher than its crystalline phase throughout the entire simulated temperature range.
|
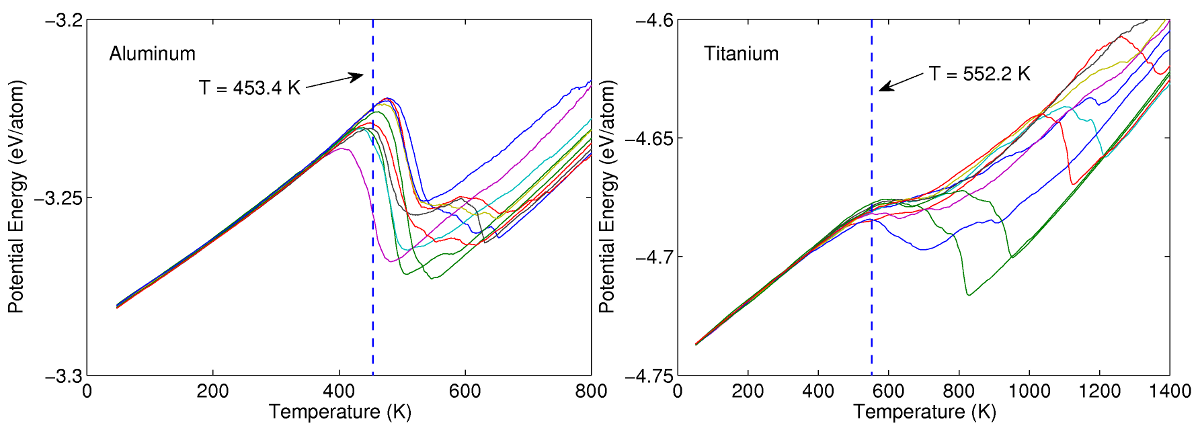 Since amorphous materials are metastable, they may recrystallize if subjected to sufficiently high temperatures. To determine the temperature at which the materials recrystallize, the amorphous phases are heated while the potential energy of the system is recorded. Since crystalline atomic configurations are more energetically favourable than amorphous states, the phase transition will be accompanied by a drop in potential energy. The recrystallization temperature is therefore determined by noting the temperature at which a sudden decrease in potential energy is observed. Ten simulations are conducted for both aluminum and titanium to ensure sufficient sampling of the statistical ensemble. Results from the MD simulations show that the recrystallization temperatures of amorphous aluminum and titanium are approximately 450K and 550K respectively.
Since amorphous materials are metastable, they may recrystallize if subjected to sufficiently high temperatures. To determine the temperature at which the materials recrystallize, the amorphous phases are heated while the potential energy of the system is recorded. Since crystalline atomic configurations are more energetically favourable than amorphous states, the phase transition will be accompanied by a drop in potential energy. The recrystallization temperature is therefore determined by noting the temperature at which a sudden decrease in potential energy is observed. Ten simulations are conducted for both aluminum and titanium to ensure sufficient sampling of the statistical ensemble. Results from the MD simulations show that the recrystallization temperatures of amorphous aluminum and titanium are approximately 450K and 550K respectively.
|
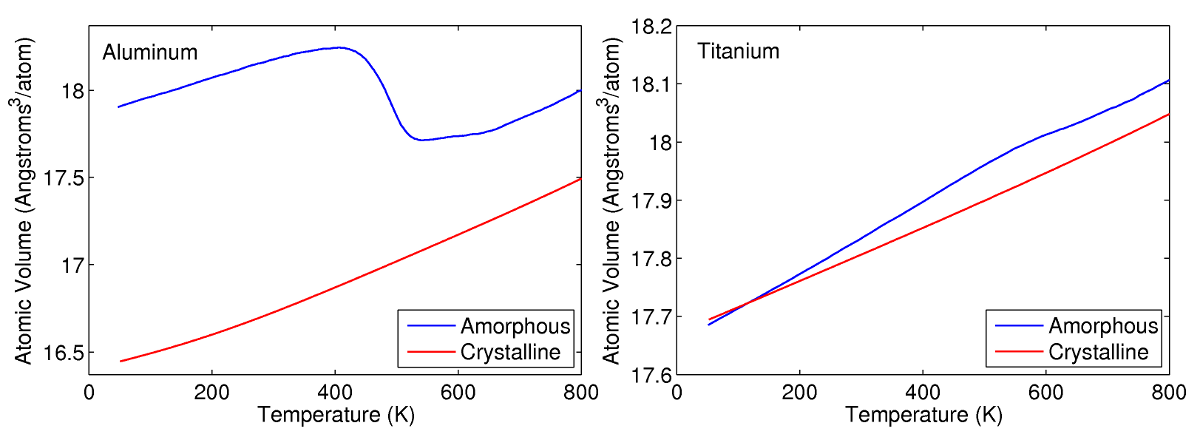 It is known that amorphous states tend to be less dense than their crystalline counterparts, and thus a change in phase will result in a negative change in volume. The amount of shrinkage that occurs during recrystallization is scarcely reported in the literature for aluminum and titanium. MD simulations are therefore used in this research to quantify their changes in volume as a result of devitrification. To accomplish this task, the average atomic volume profiles of amorphous aluminum and titanium are compared to those of their crystalline states. Using these curves, a linear expansion coefficient due to recrystallization is calculated assuming isotropic behaviour. A large negative coefficient is calculated for aluminum due to the extreme changes in volume during recrystallization. A relatively small value is determined for titanium owing to the small differences in density between its amorphous and crystalline phase.
It is known that amorphous states tend to be less dense than their crystalline counterparts, and thus a change in phase will result in a negative change in volume. The amount of shrinkage that occurs during recrystallization is scarcely reported in the literature for aluminum and titanium. MD simulations are therefore used in this research to quantify their changes in volume as a result of devitrification. To accomplish this task, the average atomic volume profiles of amorphous aluminum and titanium are compared to those of their crystalline states. Using these curves, a linear expansion coefficient due to recrystallization is calculated assuming isotropic behaviour. A large negative coefficient is calculated for aluminum due to the extreme changes in volume during recrystallization. A relatively small value is determined for titanium owing to the small differences in density between its amorphous and crystalline phase.
|
Lattice Design with Near Zero Thermal Expansion
 Using information obtained from the previous studies and an iterative process, a lattice is designed to have a CTE of zero between 150-350K, the estimated temperature range experienced by the mirrors in space. When subjected to thermal cycling, a good design should also exhibit shakedown quickly such that there is no accumulation of plastic strain between subsequent cycles. To minimize the void regions in the lattice which are optically undesirable, a nearly solid configuration is proposed. In this design, a skewness angle of 30 degrees is implemented in order to reduce the empty spaces in between unit cells and the aluminum constituents are enlarged such that only small slits remain in between the two materials. It is important to note that the configuration of the type 1 constituent has little impact on the thermal properties of the lattice, provided it behaves isotropically and only touches the constituent 2 members at the three interfaces.
Using information obtained from the previous studies and an iterative process, a lattice is designed to have a CTE of zero between 150-350K, the estimated temperature range experienced by the mirrors in space. When subjected to thermal cycling, a good design should also exhibit shakedown quickly such that there is no accumulation of plastic strain between subsequent cycles. To minimize the void regions in the lattice which are optically undesirable, a nearly solid configuration is proposed. In this design, a skewness angle of 30 degrees is implemented in order to reduce the empty spaces in between unit cells and the aluminum constituents are enlarged such that only small slits remain in between the two materials. It is important to note that the configuration of the type 1 constituent has little impact on the thermal properties of the lattice, provided it behaves isotropically and only touches the constituent 2 members at the three interfaces.
|
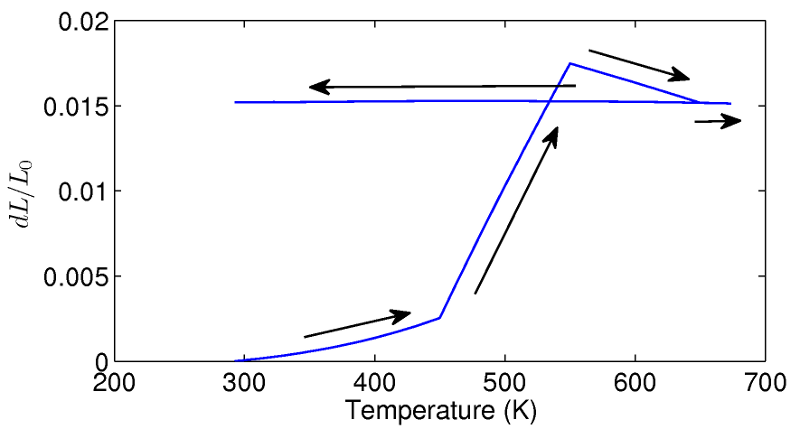 In the design of the bi-material lattice, it is assumed that the amorphous constituents will undergo recrystallization through heating. FE models and simulations are therefore used to examine the behaviour of the unit cell as the constituents recrystallize and to study the long term thermal properties of the lattice. A unit cell modeled in ABAQUS is first cycled between 293-673K to induce recrystallization. During this first heating cycle, large deformations are observed in the lattice due to the extreme changes in the lengths of the aluminum members. The recrystallization of the constituents causes the lattice to grow by approximately 1.5%.
In the design of the bi-material lattice, it is assumed that the amorphous constituents will undergo recrystallization through heating. FE models and simulations are therefore used to examine the behaviour of the unit cell as the constituents recrystallize and to study the long term thermal properties of the lattice. A unit cell modeled in ABAQUS is first cycled between 293-673K to induce recrystallization. During this first heating cycle, large deformations are observed in the lattice due to the extreme changes in the lengths of the aluminum members. The recrystallization of the constituents causes the lattice to grow by approximately 1.5%.
|
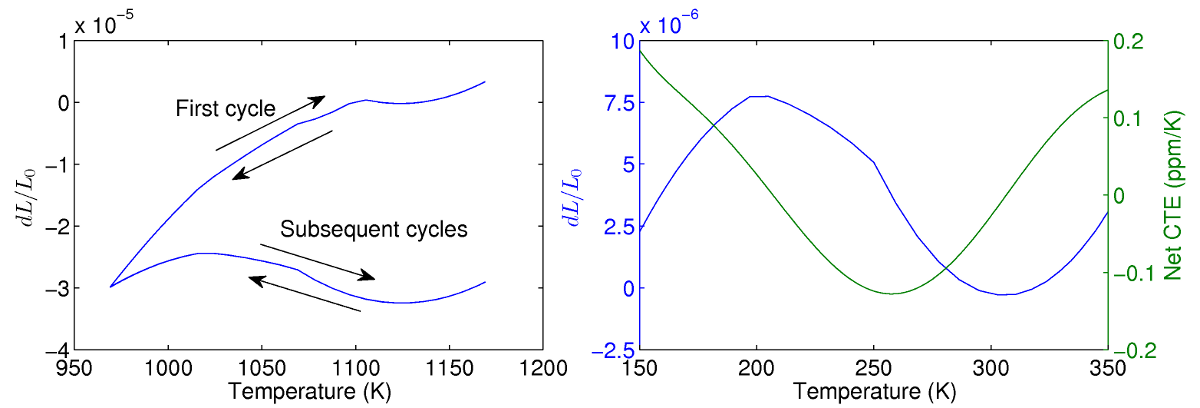 Immediately following the first heating phase, the unit cell is cycled between 150-350K for 5 iterations to examine its thermal properties. During the first cycle, further plastic deformation within the unit cell is observed as the temperature is decreased to 150K. Subsequent thermal cycles show repeatable performance however, indicating that shakedown has occurred. The stable thermal properties of the unit cell are examined in closer detail and the net CTE of the structure is calculated. It is found that the CTE of the lattice oscillates sinusoidally between roughly +0.2 and -0.1 ppm/K, and is very close to target value of zero. The sinusoidal behaviour is a result of the temperature dependent thermal properties of aluminum and titanium, which causes the ratio of their CTEs to fluctuate as a function of temperature. Thus through iterative designs, an
amorphous lattice which undergoes recrystallization and shakedown has been successfully
proposed which exhibits long term near zero thermal expansion between 150-350K.
Immediately following the first heating phase, the unit cell is cycled between 150-350K for 5 iterations to examine its thermal properties. During the first cycle, further plastic deformation within the unit cell is observed as the temperature is decreased to 150K. Subsequent thermal cycles show repeatable performance however, indicating that shakedown has occurred. The stable thermal properties of the unit cell are examined in closer detail and the net CTE of the structure is calculated. It is found that the CTE of the lattice oscillates sinusoidally between roughly +0.2 and -0.1 ppm/K, and is very close to target value of zero. The sinusoidal behaviour is a result of the temperature dependent thermal properties of aluminum and titanium, which causes the ratio of their CTEs to fluctuate as a function of temperature. Thus through iterative designs, an
amorphous lattice which undergoes recrystallization and shakedown has been successfully
proposed which exhibits long term near zero thermal expansion between 150-350K.
|